In honor of Biophysics Week and Brain Awareness Week, we asked Biophysical Society member Anna Koster, PhD student in the labs of Merritt Maduke and Justin Du Bois at Stanford University to write about her biophysics research related to brain functioning.
In honor of Biophysics Week and Brain Awareness Week, we asked Biophysical Society member Anna Koster, PhD student in the labs of Merritt Maduke and Justin Du Bois at Stanford University to write about her biophysics research related to brain functioning.
What is the connection between your research and brain functioning?
All of the electrical signals that are transmitted in the human body are mediated by a class of proteins called ion channels that sit within cell membranes and allow a variety of electrolytes (primarily sodium, potassium, calcium, and chloride ions) to selectivity pass in and out of the cell. Specialized collections of ion channels present in different tissues and organs regulate everything from your heartbeat and muscle contraction to the signals sent from your brain. Essentially, you can think of “excitable” cells as miniature electrical circuits—the cell membrane acts as a capacitor that stores charge in the form of ions on either side of the cell membrane. The difference in amount of charge on either side of this membrane capacitor, together with the chemical concentration gradient of the ions, establishes the driving force for the transmission of electrical signals in the body.
Scientists understand how these processes work at a basic level, but they involve an incredibly complex system of interactions, and there is a lot we still have to learn. My particular research project focuses on an understudied piece of this puzzle, a class of ion channels called CLC chloride channels. There are nine different subtypes of this protein in the human body, and generally speaking, they allow the passage of negative charge (in the form of chloride ions) across cell membranes. One of these, CLC-2, is highly expressed in both neurons and glia in the brain, but its function remains poorly understood. My research seeks to develop new molecular tools that will facilitate study of this ion channel both in the healthy brain and in disease states.
Why is your research important to the public?
The CLC-2 chloride channel is present in both neurons and glia in the brain. Neurons, which compose about 20% of the cells in the brain, are the electrically excitable cells that are responsible for the firing of action potentials. Glia, which are non-excitable cells, make up about 80% of your brain. People once thought these cells had only a passive structural function, but we now know that glia play a number of mechanistic roles in overall brain function and health. While we know that CLC-2 is present in the brain, much of the insight into CLC-2 function in this organ has come from selective gene knockout studies in mice. Mice lacking the CLC-2 gene exhibit severe white matter degeneration (leukoencephalopathy) over time, which suggests a critical role for CLC-2 in glial cells. In neurons, a variety of suggestions have been made regarding the role of CLC-2 in cellular excitability and ion homeostasis.
While mouse genetic studies provide clear evidence that CLC-2 has an important function in the brain, such studies are a rather crude approach to studying its specific function and behavior, particularly since genetic knockouts can cause compensatory changes in protein expression. To avoid such complications, an alternative approach is to use small-molecule tools that can rapidly and reversibly modulate CLC-2 function. For many ion channels, nature has provided such tools in the form of molecular toxins that organisms produce as a defense mechanism. One common example is tetrodotoxin (TTX), a potent neurotoxin that is found in pufferfish, ocean sunfish, and rough-skinned newts. The poison works by acting as a molecular cork in voltage-gated sodium channels, blocking the passage of sodium ions, which results in paralysis and stopping the heart of any potential predator. Chemists have isolated these naturally occurring toxins, determined their structures, and figured out how to synthesize these same molecules in the lab. In doing so, chemists are able to take advantage of the exquisite ability of these molecules to recognize and bind with high affinity to a single ion channel out of the thousands of types of proteins in the human body. In the lab, small-molecule tools like TTX allow scientists to study ion channels in a variety of ways. Such molecules can be used as structural probes of sodium channels expressed in cultured cells, or they can be used to evaluate how inhibition of sodium channels in brain tissue affects action potential firing and overall signal propagation. These molecular tools have been invaluable for studying how ion channels work at the biophysical and physiological levels.
In contrast to sodium channels, the number of small molecules that specifically recognize and bind to members of the CLC chloride channel family is extremely limited. Of the molecules that are known to bind and inhibit the flow of chloride current, most require high concentrations to have an effect and will non-specifically stick to a variety of other proteins in the body besides CLC channels. The challenge set before us as chemists was to design a novel molecular tool that would selectively and potently bind to CLC-2, in order to study its function in the brain. By employing a combination of structural biology, electrophysiology, modern computational methods, and chemical synthesis, we are developing new CLC-2 specific small-molecule leads for elucidating the role of CLC-2 in healthy brain tissue, as well as in disease states like epilepsy and leukoencephalopathy. This knowledge can provide the basis for understanding and treating a variety of neurological disorders.
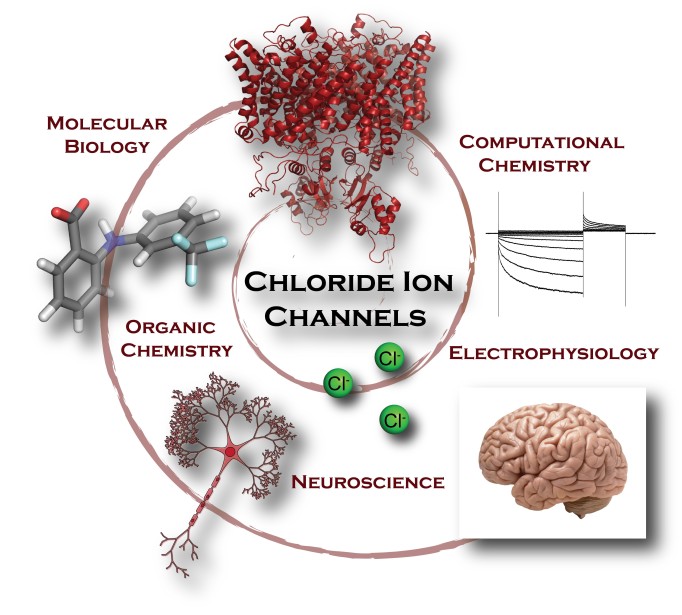
How did you get into this area of research?
I came to graduate school at Stanford with a background in organic chemistry but with a strong interest in learning to apply these skills to tackle molecular problems in the biological sphere. With the School of Medicine directly across the street from the Department of Chemistry, it was easy to establish a collaboration between the Maduke and Du Bois labs to combine their expertise in electrophysiology and CLC ion channel physiology with chemical synthesis. This collaboration, together with fellowship and grant support from a number of interdisciplinary institutes at Stanford (Bio-X, the Center for Molecular Analysis and Design (CMAD), and the center for Chemistry, Engineering, and Medicine for Human Health (ChEM-H)), enabled us to take on this challenging project at the interface of chemistry and biology. We were particularly drawn to CLC-2 because of its fascinating yet poorly understood physiology in the brain and the lack of tools available to study it.
How long have you been working on it?
We have been working on this project for approximately 4 years.
Have you had any surprise findings thus far?
CLCs have traditionally been very challenging targets, and we had very few leads to follow when we began this project. At the outset, we were working with molecules with potency towards CLC-2 in the ~1 mM range and with little to no selectivity among the human CLC homologs (and even other classes of ion channels). Employing a relatively small automated patch-clamp electrophysiology screen, we identified a first lead compound with potency in the low-micromolar range. With further structural modification and rounds of structure-activity relationship studies, we improved the potency to mid-nanomolar levels. These newly developed molecules are the most potent small-molecule CLC inhibitors discovered to-date—it was a very exciting day in lab when we first found those results! We are currently working to understand the basis for molecular recognition between these small-molecules and the CLC protein with the goal of being able to rationally design better small-molecule tools that can act as tunable modulators (either activators or inhibitors) of CLC-2 function.
What is particularly interesting about the work from the perspective of other researchers?
We believe that our research will be particularly impactful for other researchers in the field of neuroscience. We have developed the most potent small-molecule CLC-2 inhibitor known to-date. This molecule can be used as a pharmacological tool to establish the physiological function of CLC-2 in the brain. In addition, we are working to modify the molecular scaffold of our CLC-2 inhibitor to provide tools with a variety of purposes. For example, by attaching biotin and fluorescent dyes, we aim to provide a tool for imaging and visualizing under a microscope exactly where these ion channels are located and how they are trafficked within neuronal and glial cells. Altogether, the molecular tools we are developing will be vital for studying the function of CLC-2 ion channels in tissue, cells, and at the atomic level.
What is particularly interesting about the work from the perspective of the public?
Given the significant advances in science and technology, it is remarkable how little we still know about the workings of the human brain. Most of the work involving electrical signaling in the brain has focused on sodium, potassium, calcium, and protons as carriers of charge, however it is clear that chloride transport plays a critical, yet understudied, role as well. In the 26 years since the discovery of the CLC-2 chloride channel, some progress has been made in understanding its role in the brain, but many questions remain, arguably due to the absence of selective tools for reversibly modulating CLC channel function in live cells. It is our hope that the molecular tools we are developing will enable research to give us an enhanced understanding of CLC-2 in the brain and allow us to begin studying the possible role of CLC-2 in epilepsy and normal brain development. This work also has potential therapeutic impact in that the small molecules we are developing can offer prospective leads for drug development in the treatment of neurological disorders.
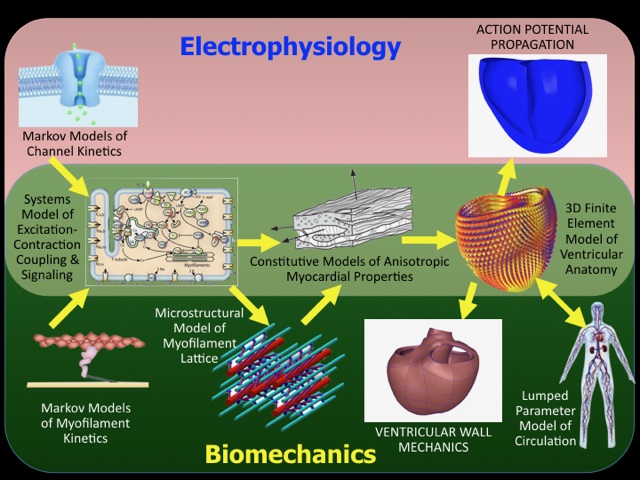
 Daniel Beard
Daniel Beard
 Andrew McCulloch
Andrew McCulloch